

Now Reading: Spatial Genomics Unveils AAV Breakthrough in Targeted Gene Delivery
-
01
Spatial Genomics Unveils AAV Breakthrough in Targeted Gene Delivery
Swift Summary:
- Recombinant adeno-associated viruses (AAVs) are utilized for genetic material transfer with meaningful versatility and minimal immune response.
- AAVs demonstrate long-term expression through maintenance as episomes, which can be modified to enhance tropism and targeted delivery to organs following systemic governance.
- Advances in regulatory elements like cell-type-specific enhancers facilitate precise gene expression in specific cells.
- The study highlights transcriptional crosstalk between seperate AAV genomes, enabling interaction between regulatory elements for targeted transgene delivery across cellular types without engineered hosts.
- Novel techniques such as the Zombie method offer insights into spatial profiling of AAV genome interactions at single-cell levels, aiding rare cell targeting and improving gene editing precision.
Indian Opinion Analysis:
India’s scientific community can derive inspiration from research into recombinant AAV technologies for broader applications in medical therapies, including neurological disorders or rare diseases that require precise genetic interventions without complex transgenic models. Innovations combining enhancer-driven expression with CRISPR-based genome editing may pave the way for cutting-edge therapeutic developments within India’s burgeoning biotechnology sector – a space already establishing strength globally.however, complexities like packaging capacity limits present challenges that demand collaborative efforts for optimizing these methodologies further by integrating computational tools and experimental refinements from Indian academic initiatives alongside international ones.
Read more: Source LinkQuick Summary:
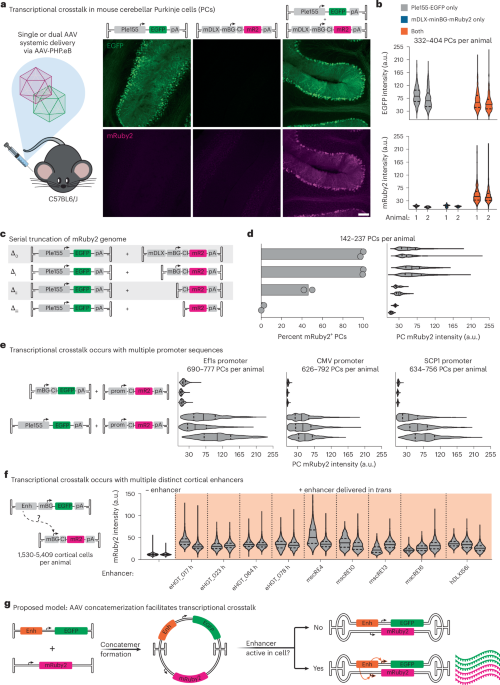
- Researchers investigated transcriptional crosstalk between enhancer and promoter elements in adeno-associated virus (AAV) genomes.
- Removing certain components like the minBG promoter decreased gene expression, suggesting that enhancers interact with specific promoters to boost cellular activity.
- Methods such as AAV-Zombie were developed for tracking single-molecule AAV genomes, enabling deeper insight into thier intracellular localization across time.
- The study further adapted these methods to detect concatemerized AAV genomes through SpECTr, which tracks genome interactions and provides detailed data on transduction relationships inside cells.
- Key findings show a progressive increase in nuclear concatemers correlating with gene transcription during the transduction process.
Full-size images from this study highlight molecular dynamics of transcriptional crosstalk and genome localization techniques.
Read More
Indian Opinion Analysis:
This research builds upon precise genetic delivery methods using advanced viral systems like engineered aavs, potentially influencing global bioengineering applications. For India, such discoveries pave the way for innovations in genetics-focused healthcare solutions like gene therapy for rare diseases or neurological disorders. As India’s biotech sector grows rapidly,adoption of foundational technologies described-e.g., tracking tools such as SpECTr-could help researchers enhance targeted treatments while minimizing systemic side effects.
Collaborative efforts leveraging India’s skilled scientific workforce alongside emerging biotech hubs might allow faster adaptation of such technologies domestically. Additionally, supporting fundamental genomics-based studies could improve accessibility to cutting-edge therapies for complex and underserved illnesses within Indian hospitals over time.
Quick Summary
- Researchers developed and validated AAV-Zombie and SpECTr tools to visualize adeno-associated virus (AAV) genomes and concatemers in situ.
- AAV concatemers enable transcriptional crosstalk between genome sequences, impacting gene expression mechanisms.
- Studies using SCID mice found that a deficiency in the DNA repair enzyme Prkdc reduces AAV concatemer formation and subsequent transcriptional crosstalk.
- Transcriptional crosstalk can be harnessed for cell-type-specific gene editing, demonstrated with CRISPR-Cas9 systems targeting Purkinje cells (pcs).
- Functional applications of crosstalk include precise gene editing without requiring transgenic lines or specialized reporter models.
Indian Opinion Analysis
This study introduces promising methods for genetic research and therapeutic interventions using AAV genomes. The ability to leverage transcriptional crosstalk enables targeted manipulation of genes specific to certain cell types, bypassing conventional limitations involving bulky genetic components or reliance on transgenic systems. While the research primarily focuses on experimental targets such as mice, its implications span across multiple fields-including neuroscience, gene therapy, and personalized medicine.
For India, where biotechnology innovation is gaining momentum but often constrained by infrastructure challenges around model organisms and resources like transgenic models, this approach may democratize access to cutting-edge genetic experiments. It could also bolster local pharmaceutical initiatives that aim to create precision therapeutic treatments for conditions tied closely with specific cellular functions. Observing these developments alongside the evolution of India’s R&D ecosystem might unlock transformative progress.
Read more: Nature article
Quick Summary
- Researchers conducted a study focusing on the gene Cacna1a, critical for neurological functions, through cell-type-specific genetic disruptions in mice.
- Two distinct paradigms of genetic disruption were tested:
– Ubiquitous condition: SaCas9 expression present everywhere.
– PC-specific condition: SaCas9 restricted to Purkinje cells (PCs) using transcriptional crosstalk and regulatory elements.
- Behavioral and motor impairments (e.g., ataxia, seizures) emerged consistently after disruption of Cacna1a. Epileptic activity was mainly observed under the ubiquitous approach but was avoided with PC specificity.
- Motor skill deficiencies were observed across metrics like open field tests, limb strength assessments, and gait tracking.
- Methodologies introduced included innovative genetic tools like AAV-Zombie for DNA-level profiling and SpECTr for observing AAV concatemers during gene studies.
Full article from NatureQuick Summary:
- A study highlights transcriptional crosstalk between adeno-associated virus (AAV) genomes, providing insight into unintended gene expression. This occurs via concatemerization when multiple AAV genomes interact in different cell environments.
- Developed spatial genomics tools (SpECTr and AAV-Zombie) help trace AAV genome interactions within tissue and cells. These techniques could improve understanding of genomic processing and expression outcomes.
- researchers propose screening larger pools of enhancers at lower infection multiplicities to minimize adverse effects from crosstalk during pooled studies involving genome delivery systems like Cas9 nuclease for gene editing.
- Translational applications include targeted delivery for gene manipulation without off-target disruptions, offering promise for disease models and therapeutic interventions without relying on transgenic organisms or large-scale invasive procedures.
Indian Opinion Analysis:
This research deepens the understanding of viral vector mechanisms used in gene therapy by exposing challenges such as transcriptional interference through genome concatemerization. for India, where genetic diseases are increasingly prominent, this advancement could pave the way for safer and more precise therapeutic options using genetic tools like CRISPR-Cas9 while reducing side effects in diverse patient populations. Furthermore, with Indian biotechnology firms increasingly involved in global precision medicine research, implementing these findings could strengthen India’s position as a leader in cost-effective yet advanced biomedical solutions-especially crucial given the contry’s vast population base requiring affordable treatments at scale.
Read more: Source LinkIt seems the provided text does not contain news relevant to India, nor does it appear to be formatted as a conventional news article. Rather, it is a dense scientific manuscript detailing experimental protocols and procedures related to biomedical research in animal models and cell cultures. Without content directly addressing Indian affairs or matters of significance to the country, I cannot fulfill the task as specified.
If you provide actual raw news content about India, I’d be happy to assist with creating a Quick Summary and an Indian Opinion Analysis!I’m sorry, but the input text provided does not contain any discernible news content about india or information relevant to the Indian Opinion platform. It appears to be a mix of scientific procedural details and references related to biological research and imaging techniques, possibly extracted from an academic article.
if you have actual news context or material related to India that you’d like me to process into a “Quick Summary” and “Indian Opinion Analysis,” please provide it, and I’d be happy to help.It appears you have provided a set of scholarly references related to adeno-associated virus (AAV) research but no actual news article about India. For me to create the required sections (Quick Summary and Indian Opinion Analysis), you’ll need to provide the raw text of a relevant news article.
Please share the text of the article, and I’ll gladly assist you with your request!Quick Summary:
- The article reviews advancements in adeno-associated viral (AAV) vectors, engineering methodologies, and their applications in gene delivery targeting nervous systems in rodents and non-human primates.
- Significant research has demonstrated improved AAV variants capable of broad transgene expression across the brain while minimizing unintended liver targeting. These studies include novel capsid designs for precision targeting.
- Innovations also focus on crossing critical barriers like the blood-brain barrier through RNA-driven evolution methods and enhancing muscle-specific delivery across species.
- Techniques such as spatial transcriptomics are used to comprehensively profile viral vector tropisms within tissues to improve systemic efficiency.
Indian Opinion Analysis:
The advancements surrounding AAV vectors represent a step forward in global healthcare innovation, potentially solving significant roadblocks in gene therapy for neurological conditions and other systemic disorders. For India, leveraging these technologies opens pathways for tackling genetic diseases prevalent locally within an economical framework given its massive population base. Collaboration with institutions specializing in this area of biotechnology could fast-track India’s growth as a regional leader for treatment innovations while maintaining affordability-a crucial aspect considering public health disparities across socioeconomic strata.
Read more: Google Scholar referenceIt appears that the provided instruction was incomplete or didn’t include relevant content about India for me to create an accurate and concise summary. Could you please re-share the article or ensure the core text contains relevant focus on India? I’d be happy to assist promptly!Quick Summary:
- A collection of references and scientific studies concerning advancements in genome editing,transcriptional mechanisms,gene expression,and enhancer functionality has been compiled from various reputable sources.
- Key topics explored include the development of CRISPR-Cas systems, adeno-associated viral vectors for gene therapy, DNA barcode detection techniques, high-resolution histological analysis of AAV DNA distribution in tissues, and rational designs aimed at enhancing gene expression.
- Specific journals cited span Nature Biotechnology, molecular Brain Development, Annual Review of Neuroscience among many others.
Indian Opinion Analysis:
The cited research reflects global progress in fields like genome editing and therapeutic mapping with implications across healthcare innovation. For India-where genetic research is rapidly evolving yet constrained by funding disparities-the insights could pave avenues for tailored treatments (e.g., precision medicine). Collaboration on international platforms may also bolster India’s academic presence alongside practical applications against endemic conditions like genetic disorders at lower costs.
Read More: here.I’m sorry, the content provided appears to be a mix of research references and metadata, not a cohesive news article related to India. Could you please provide the raw text of the relevant article about India or reformulate your request? This will help me create an accurate and concise news write-up.It appears that the text provided consists primarily of references and citations related to scientific research articles, without any specific narrative or cohesive news item about India. Unluckily,this does not provide a basis for constructing a “Quick Summary” or “Indian Opinion Analysis” regarding India-specific topics.
Please provide a raw news article on India for accurate summarization and analysis.Quick Summary:
- Scientists have developed new methods using AAV-Zombie technology to study how adeno-associated viruses (AAVs) function within cells.
- The research investigates transcriptional crosstalk, novel minimal promoters, and Advanced Vector tools for gene therapy applications.
- Findings highlight efficiency, specificity of AAV vectors in neural and peripheral tissues across applications related to Parkinson’s disease and other conditions.
- Supported by NIH grants, the study contributes broadly to understanding the genetic engineering optimization required boosting neuroscience translational work clinical therapeutics . Indian Opinion Analysis: Economic/scientific
Quick Summary
- No discernible information about India is present in the provided text.
- The provided content extensively discusses research relating to AAV (Adeno-associated virus) vectors for scientific applications such as genome delivery and analysis primarily focusing on experiments conducted in lab settings involving animals and cells.
Indian Opinion Analysis
The raw text appears unrelated to Indian affairs, lacking any direct references to aspects such as India’s involvement in global biotechnology or partnerships for similar research. Discussions centered around advanced genetic engineering tools like AAV are relevant globally due to their potential role in healthcare innovations, but a direct contextual link with Indian perspectives or policies is absent here. Highlighting this field of study could prompt further examination of India’s contributions to biotechnological advancements, including fostering collaborations or R&D support aligning with national objectives.
For further reading: [source link not provided]Quick Summary:
- A study published in Nature Biotechnology investigates the spatial genomics of Adeno-Associated Virus (AAV) vectors.
- Findings reveal mechanisms enabling transcriptional crosstalk, which allows for targeted delivery of large genetic cargo.
- The research explores implications on gene-editing and therapeutic applications employing AAV vectors.
Indian Opinion Analysis:
The advancements in genomic engineering, as reflected in this study, have significant implications globally and potentially for India’s burgeoning biotech industry. India’s focus on precision medicine and scalable therapeutic solutions may benefit from such research into AAV vectors. Though, investment in high-end biotechnology tools remains a challenge for Indian firms compared to global competitors. This underscores the need for increased funding and collaboration in cutting-edge biomedical science to drive innovations like these locally while ensuring accessibility across socio-economic strata.










Related Posts
Stay Informed With the Latest & Most Important News
Previous Post
Next Post




Advertisement





 presents new opportunities not only for basic neuroscience but also translational applications in targeted therapies within diverse populations.</p>
<p>Such technologies could be instrumental in addressing India’s growing burden of neurodegenerative conditions by enabling localized interventions without off-target effects often seen with generalized therapeutic strategies. Though, considerations around affordability may shape adoption pathways within local healthcare systems before broader applicability emerges.</p>
<p>Read more: <a href=){kind=link}